Why are drugs so disappointing?
No, this isn’t an extended post about reefer madness. It’s about that horrible gap between the excitement of a new therapy for an illness, and the results that eventually come out years later.
One of my main interests is in explaining data to people in a way that is simultaneously clear and honest. The most disappointing aspect of this is that, often, things are much less exciting than they first appear. Nowhere is this more true than in the effects of much-hyped new drugs.
There are, of course, good reasons why treatments that are much talked about under-perform in subsequent trials. For example, regression to the mean, materials intended to promote investment (aka dodgy marketing), and the media’s insatiable hunger for a good story. We could also counter with the argument that we shouldn’t really expect a pill going through our digestive system or a few millilitres of liquid to change the underlying biology of a complex organism.
This isn’t everything, though.
It would be one thing if these disappointments just demonstrated that modern drugs are ineffective, but there is more to it than that; modern treatments are generally pretty impressive. For many of these treatments, a proportion of patients experience dramatically good outcomes, and I don’t just mean those patients sitting on the tail of a distribution of effects. I mean a substantial proportion of patients. In addition, modern therapeutics developed and tested in precisely the same way as these disappointing drugs can have an amazing effect. Modern treatments for HIV, as an example, are simply astounding.
So, what’s going on? How can we get better drugs, or more from the drugs we have?
There are a few things to look at. Here are just three:
- Not all diseases are diseases
- We often measure the end of the causal chain
- People vary
These are really three facets of a single problem:
Our health is influenced by a complex mixture of things. We don’t always know what these things are, so we make guesses and group people together in ways that seem sensible at the time. When we do this, we often get it wrong. This stops us using drugs efficiently.The solution, I will argue, is to model disease right from the beginning, and to update our models as new data becomes available. This relies on good quality record keeping about patient experiences.
Let’s look at the problems.
Not all diseases are diseases
This may seem like a bit of an odd statement, but it’s true. Historically, new diseases are often ‘discovered’ by someone observing that multiple patients present with similar clusters of signs and symptoms. We then end up trying to work out what caused those signs and symptoms across the group of individuals.
Sometimes, we get lucky and the people presenting with similar signs and symptoms do, indeed, share a single underlying cause (known as an aetiology). A good, relatively recent, example of this is AIDS (Acquired Immuno-Deficiency Syndrome), although even AIDS can be caused by at least two types of HIV. The history of AIDS gives a real insight into the way we fumble around trying to work back from clinical and epidemiological observations to basic biological processes.
Where we have a single underlying aetiology, we can be reasonably sure that a treatment will show some consistency of effect across patients, as shown below.
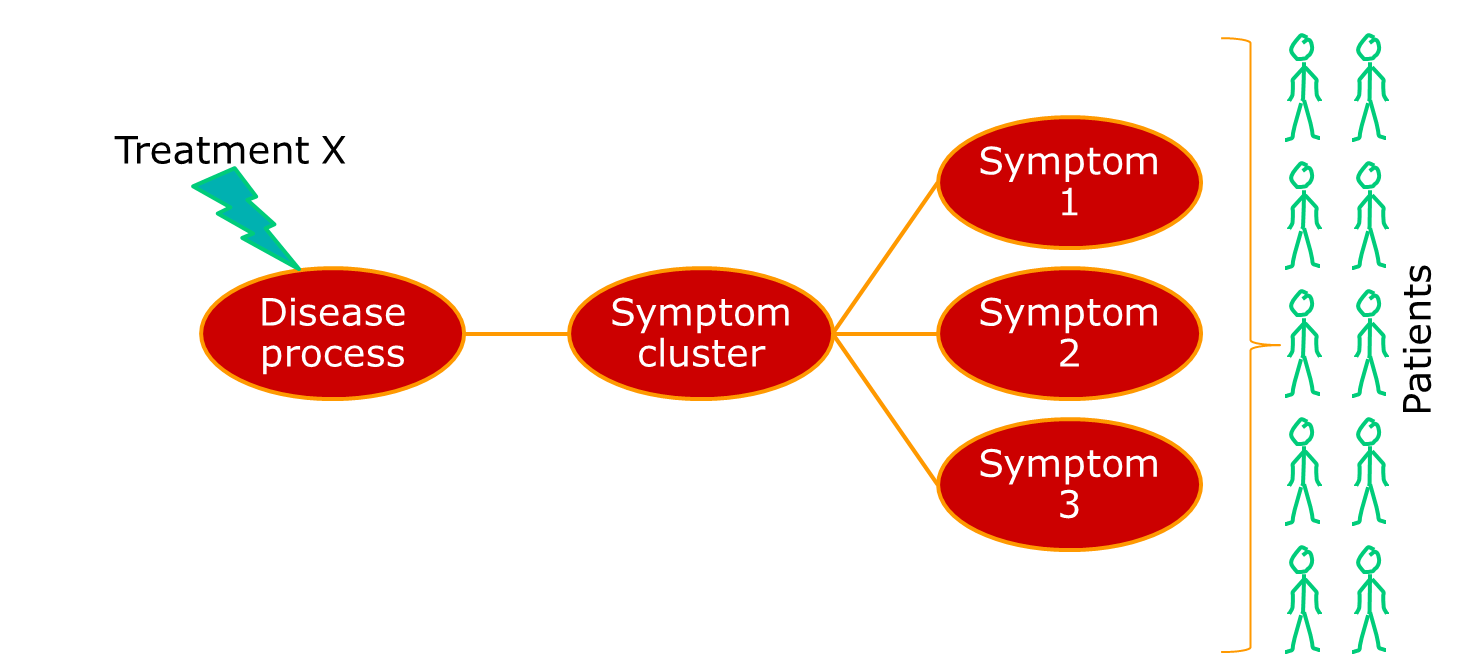 Often, though, the group of people we identify happen to show similar patterns of illness, but have very different aetiologies. A very current example of this is breast cancer, which has recently been described as being a number of separate diseases. Other examples include conditions such as non-Hodgkin lymphoma, which has been a source of nosological confusion for most of my career. The term for a pattern of symptoms that can have multiple aetiologies is a syndrome, not a disease.
Often, though, the group of people we identify happen to show similar patterns of illness, but have very different aetiologies. A very current example of this is breast cancer, which has recently been described as being a number of separate diseases. Other examples include conditions such as non-Hodgkin lymphoma, which has been a source of nosological confusion for most of my career. The term for a pattern of symptoms that can have multiple aetiologies is a syndrome, not a disease.
Therefore, not everything that we think of as a disease is a disease. Lots of them are syndromes.
The point is this: complex systems can produce the same behaviour or fault many different ways. We don’t know everything, so we have to guess. If we guess the cause of the fault incorrectly, our treatment won’t work. It might even have the opposite effect to the one we want.
The consequence of this is that, if we apply a single treatment to all the patients with a syndrome, we will only see a positive response in a subgroup. If we use average estimates of treatment effect, our treatment will look rubbish even if it works.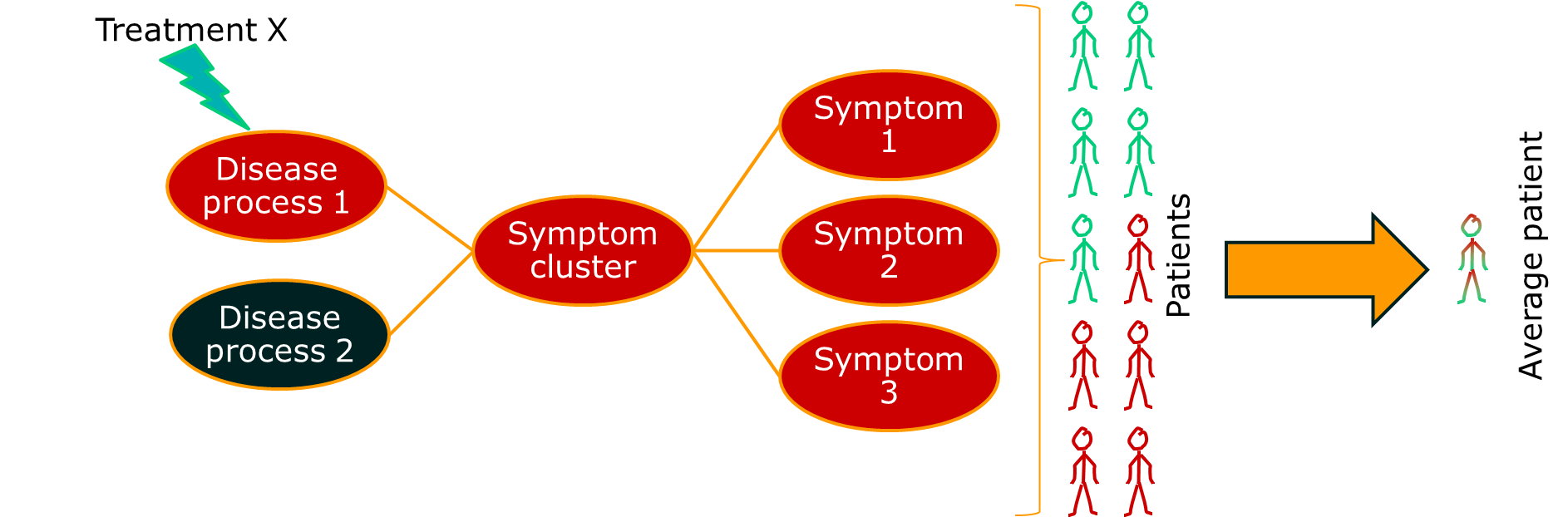
We often measure at the end of the causal chain
This is a bit of a kicker. Clinically, the aim of your treatment may be to delay death, avoid a disease exacerbation, or slow deterioration in everyday activities. However, the treatment itself has to get into your body, get to the site of action, interact with biochemical structures on the surface of cells (or whatever), cause a change in biological activity resulting in a change in the disease process and improved patient outcome. That’s a lot of things that have to happen. Add to that the observation that outcomes like death can be caused by all sorts of things, and you have a problem.
It’s not quite accurate, but the way I think of it is like trying to measure a person’s height by counting how often they bang their head on doors. It’s not great. Having said that, we can’t deny that the clinically relevant outcomes are what we should be basing clinical decisions on.
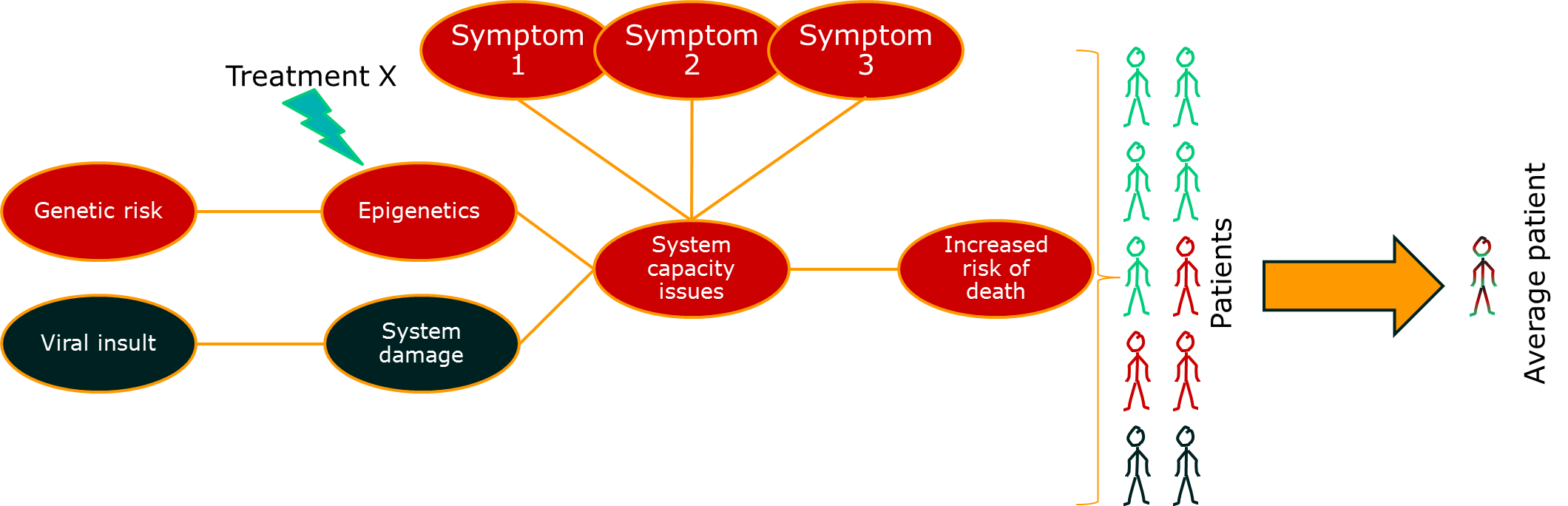 One approach to dealing with this problem is to look at some of those intermediate measures. If you can measure the activity of a drug directly, you may be able to distinguish those patients that are benefiting from those that are not. For outcomes like death, this means that you can reduce some of the impact of the long causal chain on your ability to measure what’s going on. More importantly, it gives you a chance to change treatments before it’s too late.
One approach to dealing with this problem is to look at some of those intermediate measures. If you can measure the activity of a drug directly, you may be able to distinguish those patients that are benefiting from those that are not. For outcomes like death, this means that you can reduce some of the impact of the long causal chain on your ability to measure what’s going on. More importantly, it gives you a chance to change treatments before it’s too late.
So far so good. There’s something else though. This issue relates to the issue of diseases being syndromes. Some of the variability in the big causal chain between treatment effect and outcome is due to differences in the disease process for groups of patients. If we put the two together, we might start to see the beginnings of a disease model:
 This is something that drug developers are doing already, and there’s a lot of excitement about it because is helps explain the basic science of a treatment alongside the clinically important outcomes. In other words, it helps us test the data against a consistent set of claims across the drug development process. A good example of this is some of the drugs currently getting early approval in oncology based on really very early evidence through fast-track regulatory systems. These are usually so-called targeted therapies that are designed around known disease pathways, and can show dramatic patient benefits in a specific subgroup of patients. The approach is great, although the early licensing of therapies is really only happening because there is a real, urgent, clinical need.
This is something that drug developers are doing already, and there’s a lot of excitement about it because is helps explain the basic science of a treatment alongside the clinically important outcomes. In other words, it helps us test the data against a consistent set of claims across the drug development process. A good example of this is some of the drugs currently getting early approval in oncology based on really very early evidence through fast-track regulatory systems. These are usually so-called targeted therapies that are designed around known disease pathways, and can show dramatic patient benefits in a specific subgroup of patients. The approach is great, although the early licensing of therapies is really only happening because there is a real, urgent, clinical need.
People vary
Of course, people vary. Even within one subgroup of patients, there may be people whose bodies break down drugs in a different way. Outside of trials, people may be taking all sorts of additional treatments or have other illnesses (known as comorbidities).
There is a point where individual variability and disease subgroups start to overlap. If we look at non-small cell lung cancer, there are currently genetically defined subgroups in at least three different genes, each of which has a number of more specific genetic subtypes. Furthermore, the specific genotype of the tumour may change in response to therapy (although the patient’s base genetic make-up does not change).
As we identify more and more potential subgroups, we start to come across a problem. Given that the causal pathways in illness are, as mentioned before, pretty long and we can have subgrouping at any stage along them, we get a problem of permutations. Essentially, each person could be a member of more than one subgroup, and those subgroups may interact in complex ways. Pretty soon, we end up in a situation where nearly every patient has a different, complex, subgroup background.
This third issue, then, is really a combination of the previous two. We have a long and complex causal chain and the conditions we currently understand as single diseases may be caused by a variable set of risk factors and other influences along that causal chain. Some of these factors (including genetic factors) may not even be constant over time.
How complex is that?
Pretty complex, as it happens. Usually, we don’t really know how many causal links there are in our aetiological chain, but we can model it out.
Suppose we have a nice simple disease with just four links between our disease presentation and our most basic disease cause. Suppose each of those links has four disease-relevant subgroups. This is actually quite a simple example, and would result in 256 different subgroups (44).
The thing is, if we keep subgrouping and the subgroups can overlap, eventually we have individualised medicine. This is medicine where a therapy is tailored to the individual needs of a patient. It’s been talked about for ages as a sort of holy grail of modern medicine, but it has some pretty major issues. First of all, we know relatively little about the causes of a lot of diseases. Secondly, testing the interaction between individual patient factors and outcome using traditional trial-based approaches is simply not (currently) practical.
The way this effects our current question is this. If a fairly simple condition can reasonably have 256 subgroups, and those subgroups can respond differentially to our treatment, our treatment effect is going to be pretty well hidden in the mess and noise associated with the uncertainty surrounding the condition itself.
Disease modelling
A lot of what is talked about when people discuss personalised medicine is rather trivial. Obviously, if we know the detailed aetiological background of a condition and we have a way of measuring the key variables on that causal chain, we can have a good guess at how an individual might respond to treatment. If we can group people in a way that allows us to conduct trials, we may even be able to get a clinically meaningful version of personalised medicine through regulatory authorities and the people (insurance companies and health bodies) that pay for drugs. If we do it with enough style, panache, and flair, we may even be able to demonstrate the value of this to doctors and (through them) patients.
What I’ve been introducing is not quite this.
Remember that I said that often new conditions were identified because signs and symptoms appear to cluster together in groups of patients. That is where we have to start in the real world. With no direct knowledge of aetiology, with no understanding of how a particular treatment interacts with that aetiology, and with no way of doing personalised medicine properly.
The drug development process is not set up for this situation. However, clinicians are. Or, rather, some clinicians are. They have an understanding of physiology, they may have some understanding of the biochemistry underlying the condition, particularly if they are specialists and/or spend time talking to the allied health professionals and assorted scientists floating around any decent health system. Furthermore, they are in a perfect position to observe patients, to talk with them, and to identify patterns that can be investigated and built upon.
When I talk about disease modelling, this is the basis of what I have in mind. If we return to our diagrams, it is possible to use statistical methods like factor analysis to look into how many different disease processes may underlie a complex set of signs and symptoms. Similarly, we look at the way symptoms collect together in a population of people and use that information to test theories about the structure of disease.
In the past, we relied on the observation skills and experience of individuals to understand illness structure and develop hypotheses about causation. Now, we can use technological approaches, such as centralised data sets and anonymised data mining to extend that approach.
Patient information systems are important, not just because they offer the opportunity for patients to move house regularly without all their records disappearing (a significant problem for those with chaotic lives, who are often most vulnerable), but because they may just allow us to start looking at the complex patterns of experience that indicate what is going on with disease.
That’s really my conclusion. New treatments can end up appearing disappointing. Sometimes they are but, when we see a pattern of disappointing results from new therapies, it may just be telling us something very interesting about disease. If we pick up on these clues, we may just be able to get more out of the treatments we already have and push our understanding of health and disease on just a little bit further.
Disease modelling will be a major theme of these posts. This is just an introduction to why I think explicit disease modelling is important. I hope to be able to show you some concrete examples using real diseases in the months to come.
Leave a Reply
You must be logged in to post a comment.